The Magmatomix/UMD package performs post-processing of molecular-dynamics simulations. It is best suited for ab initio calculations. It is written in python, and it is open-source. We welcome contributors and collaborators!
The package can be downloaded from Github
And if you use the code, please cite it using:
R. Caracas, A. Kobsch, N. V Solomatova, Z. Li, F. Soubiran, J.A. Hernandez, F. Soubiran. Analyzing melts and fluids from ab initio molecular dynamics simulations with the UMD package, JoVE, 175, e61534 (2021).
A new updated publication will come up soon.
UMD file format
The various parsers extract only the relevant thermodynamic information and atomic trajectories from the output of the first-principles/atomistic simulations. This info is writteni in “UMD” files. The umd files are ascii files; typical extension is .umd.dat but not mandatory.
Each physical property is expressed on one line. Every line starts with a keyword, followed by the numerical value(s), and the unit. In this way the format is highly adaptable and allows for new properties to be added to the umd file, all the while preserving its readability throughout versions.
Then each atom is detailed on one line, where
- columns 1-3 give the atomic positions expressed in reduced coordinates
- columns 4-6 give the atomic positions expressed in cartesian units
- columns 7-9 give the real Cartesian positions that take into account diffusion,
- columns 10-12 give the atomic velocities
- columns 12-15 give the atomic forces
- column 16 gives the atomic charges
- column 17 gives the atomic local magnetic moment.
All the functions related to the reading and printing of the umd files are combined into one python library, umd process.py. These functions are consistently called by all the scripts of the package after proper loading.
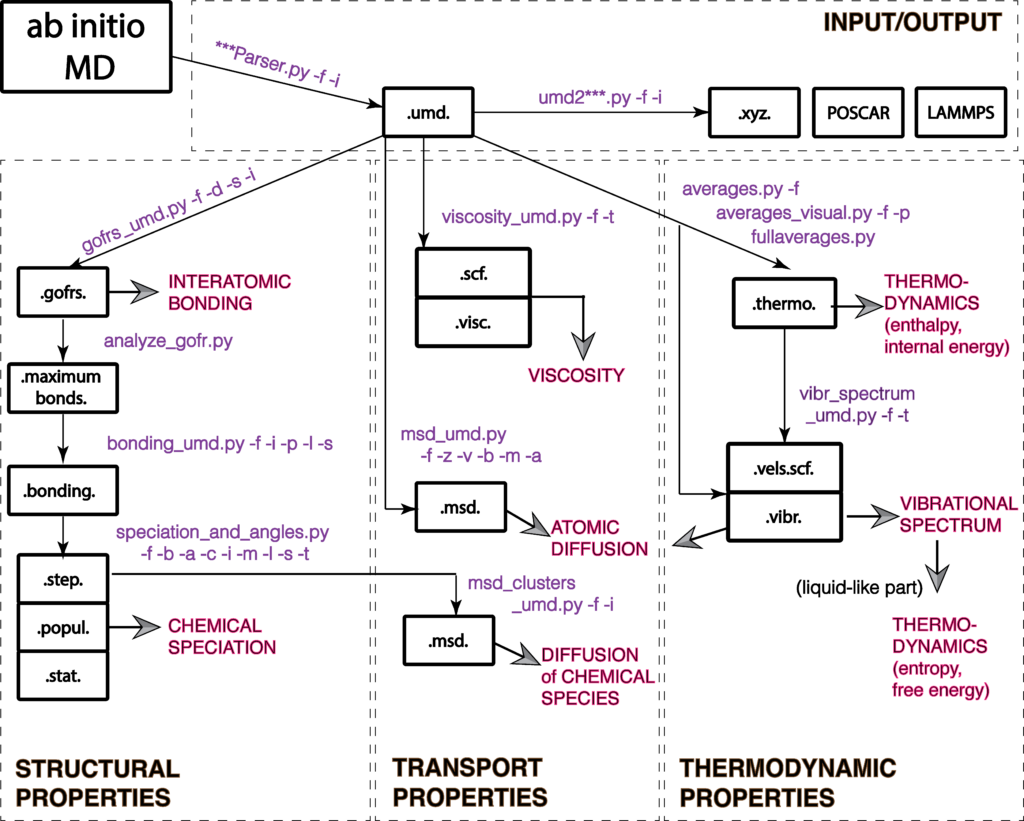
Magmatomix/UMD workflow
The following figure shows the updated workflow of the Magmatomix/UMD pacakge.
You can perform four types if analysis:
- Input/output to and from ab initio packages, like VASP or Qbox into umd, xyz, etc files
- Structural analysis, like interatomic bonding, chemical speciation, lifetimes of clusters, liquid vs vapor identification, interatomic angles
- Transport properties like diffusion coefficients and viscosity
- Thermodynamic analysis, like internal energies, heat capacity, vibrational spectrum
SHORT TUTORIAL
The hands-on session can now use docker as a container where you can run the entire magmatomix/umd package. Though it is slow, it works! For this, run the following steps:
- First gather the data.
Create a folder in your home directory that you can name, for example:
UMDschool
- Download an example file, for example this file:
https://github.com/rcaracas/UMD_package/blob/master/pyrolite.melt.a12.T4K.umd.dat
and put it in the UMDschool folder. Using the terminal window (command in windows) go to this directory and check that the file exists where it should be. Use ls or dir to check for its existence.
- Second, install docker for desktop. Take it from:
https://www.docker.com/
- Then, open the docker app
- Go back to the terminal (command) window, in the UMD school directory and type:
docker pull tbogels/docker_umd:latest
This starts the docker setup and loads the container, but it is not running it yet.
- Then type (to run the docker):
in linux/mac:
docker run -it --name docker_umd -v $PWD:/app/data tbogels/docker_umd /bin/bash
in windows 11:
docker run -it --name docker_umd -v %cd%:/app/data tbogels/docker_umd /bin/bash
in windows with PowerShell:
docker run -it --name docker_umd -v ${PWD}:/app/data tbogels/docker_umd /bin/bash
In your terminal window, where you have started the docker container, the docker itself moved the files you previously had into a new virtual folder called “data”. Go there, using:
cd data
The rest of the exercises will be done in this folder. You can run any script of the package using the Python3 from inside the docker container (not the one you might already have on your computer). You would typically run the scripts as:
python3 ../the_name_of_the_script_you_want_to_run.py
Note that all scripts come with the option -h, which gives you a brief help file, a few lines explaining how to use the script, and what each flag means and does. Don’t hesitate to use it!
For example, you can extract some thermodynamic average values like:
python3 ../averages.py -f pyrolite.melt.a12.T4K.umd.dat
or, you can transform the umd file into a xyz file, that you can use later for visualization purposes:
python3 ../umd2xyz.py -f pyrolite.melt.a12.T4K.umd.dat
Note that -f is the flog for the file name of the umd file.
If you want to use the speciation script, you must define the bond lengths, i.e., the radius of the first coordination spheres around each atom. You can either do that using the analyze_gofr.py script, but in the docker, this would not work. Then, you can use, for example, these values:
Na O 3.7
Ca O 3.5
Fe O 2.9
Mg O 3.0
Al O 2.8
Si O 2.45
O O 1.7
Just copy and paste them into an ascii file, for example “bonds.inp”
- For Visualization, we prefer to use VMD. Download and install it from:
https://www.ks.uiuc.edu/Research/vmd/
Please note that you might need to register to use it.